We are happy to announce that our latest work on the modular flow design for meta-selective arylation of anilines was published earlier this week in Angewandte Chemie International Edition (DOI: 10.1002/anie.201703369). Today, we want to take you behind-the-scenes on how the project came into existence, what the major bottlenecks were along the way, and which results lead us to the breakthrough of this publication.
Bigger, better, faster?
The saga started in the late summer of 2015 when Kirsten Verstraete, a graduate student from UHasselt Belgium, arrived in our lab. Not knowing what her graduation work would entail, I presented to her the landmark paper published by Gaunt et al.[1]. The paper describes an elusive transformation: the copper-catalyzed meta-selective arylation of protected anilines, which apparently neglects all conventional rules of electrophilic aromatic substitution chemistry (Figure 1).
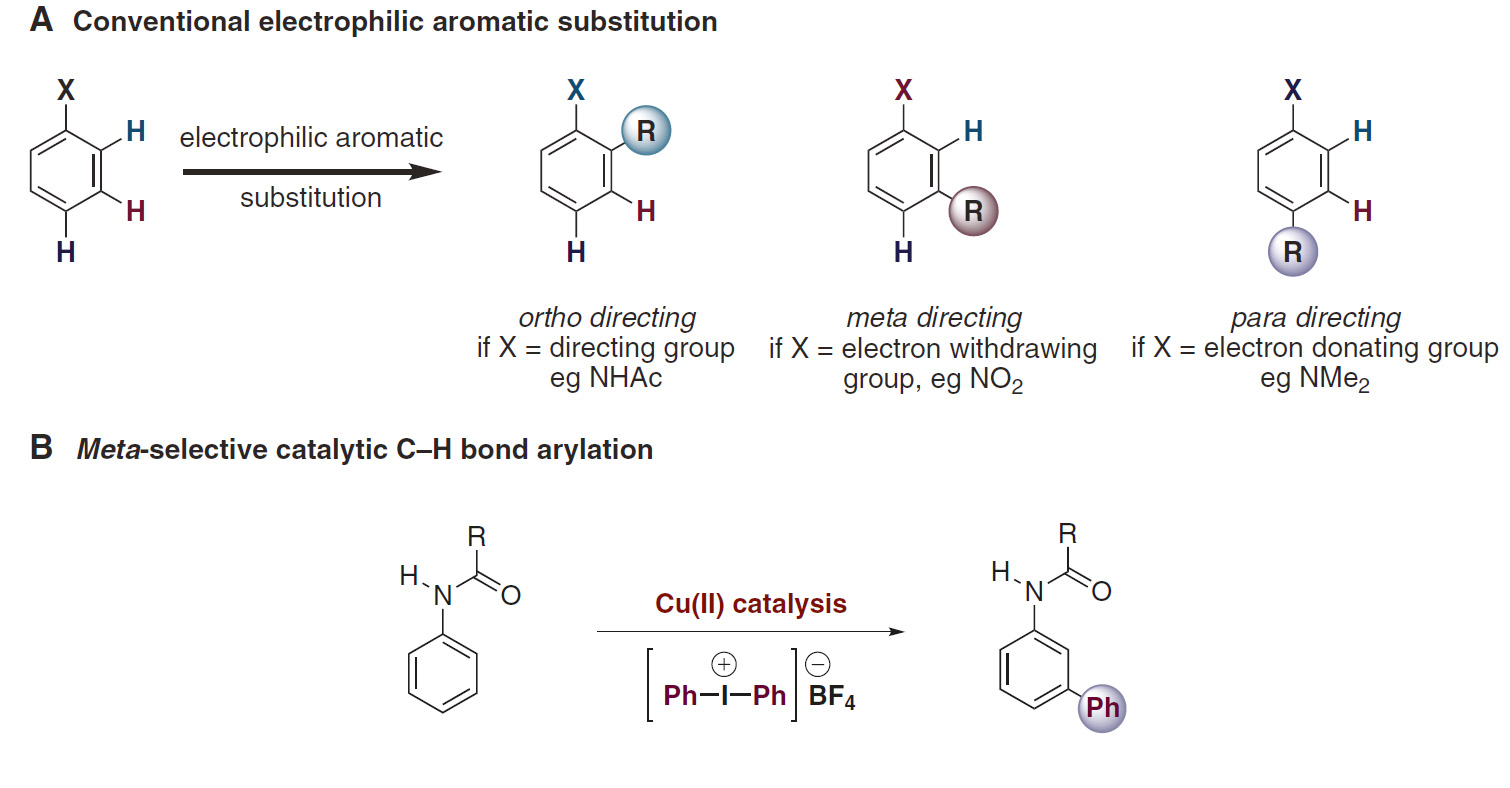
Masking my doubts with enthusiasm, I explained to her that we could make this transformation better, faster and broadly applicable. I told her (still full of enthusiasm) we would tackle all the limitations in order to give way to an efficient, cost-effective and practical process which would yield us direct access to valuable meta-arylated anilines in gram scale.
From trial to flow.
Our initial hypothesis was that the copper source (i.e. copper triflate = Cu(OTf)2) used by Gaunt et al. was perhaps not the only option as a catalyst for this transformation. The reaction is believed to proceed via an electrophilic metalation on the aniline substrate.[2] Cu(I) species are oxidized to the Cu(III)-aryl intermediate, a highly electrophilic intermediate that can undergo Heck-like meta-arylation on the aniline (Figure 2). Gaunt speculated that the active Cu(I) species were formed in situ via reduction and or disproportionation of the copper triflate. However, looking at the reaction conditions, we concluded that the use of metallic Cu(0) could be an alternative choice. Apart from being inexpensive, readily available and air stable, we hypothesized that the catalytically active species (i.e. Cu(I)) could more easily be formed from Cu(0) in the presence of highly electrophilic diaryliodonium salt.
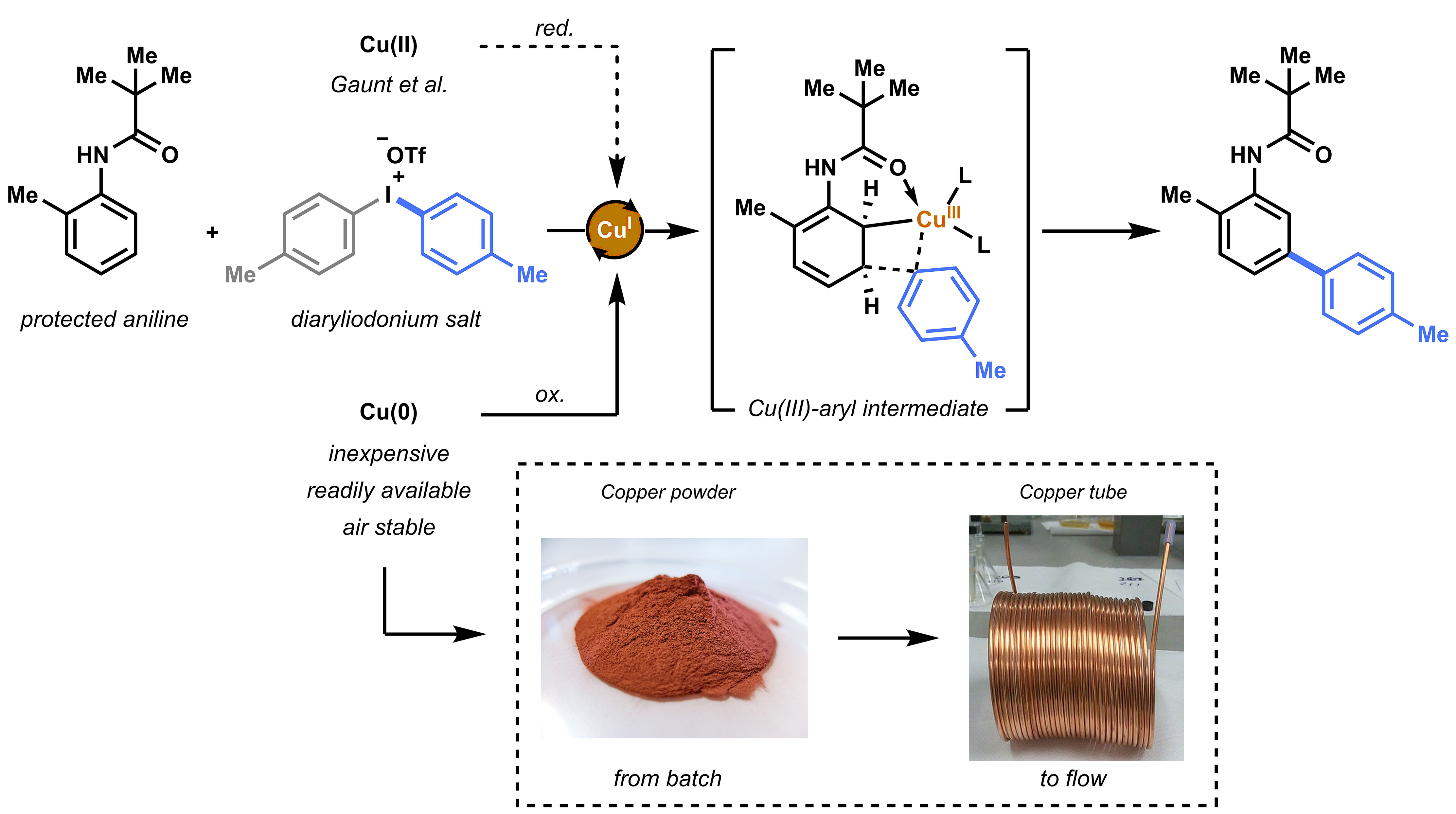
We started the initial investigation by conducting parallel batch reactions with either a Cu(II) or Cu(0) catalyst source. By checking the first GC results we knew we hit the jackpot. Not only was the Cu(0) (in this case inexpensive copper powder) suitable as catalyst precursor, but also a significant acceleration was observed. Unlike Cu(OTf)2, that needed 24 hours to complete conversion, copper powder resulted in reaction completion within 2 hours.
We speculated that this reaction could further benefit from using the so-called copper tube flow reactors (CTFRs)[3], which would allow for a significant breakthrough in operational simplicity for C−H activation chemistry. A homemade 20 mL CFTR was constructed from general purpose 1/8” copper tubing. Such tubing is normally used as gas lines for gas chromatography and could be readily ordered for less than 4 euro per meter. Using the flow reactor, full conversion and an improved yield (88%) was obtained within 20 minutes residence for our model substrate.
Homemade reagents.
After initial success with the copper tube flow reactor, it became clear to us that the diaryliodonium salts (that are used as the arylating reagent) were bringing in some limitations to the process. Despite being shelf-stable, nontoxic and synthetically useful, diaryliodonium salts continue to be limitedly available and very expensive. However, by trying to synthesize these reagents ourselves in the lab, we understood instantly why they are often neglected for application purposes. The preparation turned out to be cumbersome since hazardous reagents such as triflic acid (a super acid) and meta-chloroperbenzoic acid (a strong oxidant) needed to be handled in large amounts. On top of that, the synthesis was characterized by a huge release of energy, resulting in uncontrollable heating of the reactor at gram scale production. It was obvious that if we wanted to create a library of such compounds, this procedure needed some proper reevaluation and design.
At that point (in the late spring of 2016), Gabriele Laudadio arrived from Pisa University in Italy to join our team as a Ph.D. candidate. Enthusiastic and courageous of working with these hazardous reagents, Gabriele soon developed a preliminary capillary flow reactor and gave it a go. Substrates, acid, and oxidant were all fed separately to the flow reactor via syringe pump in order to guarantee maximum safety during operation. However, tides turned when on the first run the capillary started clogging severely and the process needed to be stopped immediately. Unable to recuperate the clogged tubing, we reconstructed a second reactor. Anticipating that under sonication clogging could be prevented, Gabriele ran to the other side of the lab to ‘borrow’ the ultrasonication bath of our colleagues. The reaction coil was submerged in the bath and subjected to ultrasound. Nicely, no reactor clogging was observed and the reaction ran smoothly. Finally, after some further optimization, the reactor could readily produce grams of diaryliodonium salts for us in a time-efficient fashion (up to 3.8 g/h). A library of diaryliodonium salts was made in in no time (Figure 3).

Copper removal within EU standards approval.
Having obtained the required diaryliodonium salts from our first module. Gabriele and I started to work on the scope of the meta-arylation reaction in flow and we obtained a total of 23 compounds. In particular, the complete scope could be conducted with only one self-made copper coil without any loss of reactivity. Even better, when conducting the reaction on gram scale, nearly quantitative yield of product was obtained. This was unprecedented. This fact in combination with the fast reaction conditions obtained in flow (20 minutes) highlighted the potential of these CTFRs to enable C−H activation chemistry for gram scale drug manufacturing.
However, we were expecting that some copper leaching would take place when running the meta-arylation reaction. Indeed, after proper analysis, we realized that a significant copper content (around 4720 ppm) was present in our reaction mixture. To tackle this problem we constructed a 5 mL extraction coil which was attached to the end of the CTFR. This way, the organic phase exiting the CTFR was merged with an aqueous ammonia solution capable of extracting the copper. The performance of the extraction could be easily verified by the naked eye since copper complexation with ammonia resulted in a deep blue color of the aqueous phase (Figure 4). To make the extraction step directly coupled with an inline phase separation, a commercially available Zaiput liquid-liquid membrane separator was attached to the extraction coil which resulted in a perfect separation of the two phases. Satisfyingly, analysis of the extracted organic phase revealed that 97% of copper was extracted with a single pass through the module. This corresponds to a residual 14.3 ppm of copper, confirming to EU norms on the limits for parenterally administered pharmaceutical substances.[4]

Why wouldn’t it hydrolyze?!
The last problem to circumvent was the flow deprotection of the amide function in order to obtain, finally, our valuable meta-arylated anilines. This turned out to be more problematic than we first anticipated. Because of the inert nature of our protection group (pivalamide), hydrolysis proved to be extremely challenging. After trying many different conditions (feel free to explore our supporting information on that matter J) no presentable results were obtained. Weeks later and about to give up on this endeavor, our last hope was the use of a 50:50 HCl:dioxane solvent mixture to further boost the deprotection procedure. Happily, our compound could be deprotected under such conditions. Translating our procedure to flow, we were thrilled to see that the pivalic protection group could be easily cleaved within only 40 minutes using superheated flow conditions (aka using a solvent above its boiling point). Such conditions are often discouraged in batch, but in flow this environment can be readily obtained by using the so called back pressure regulators. Thus, in this fashion, a series of desired meta-arylated anilines could be readily deprotected.
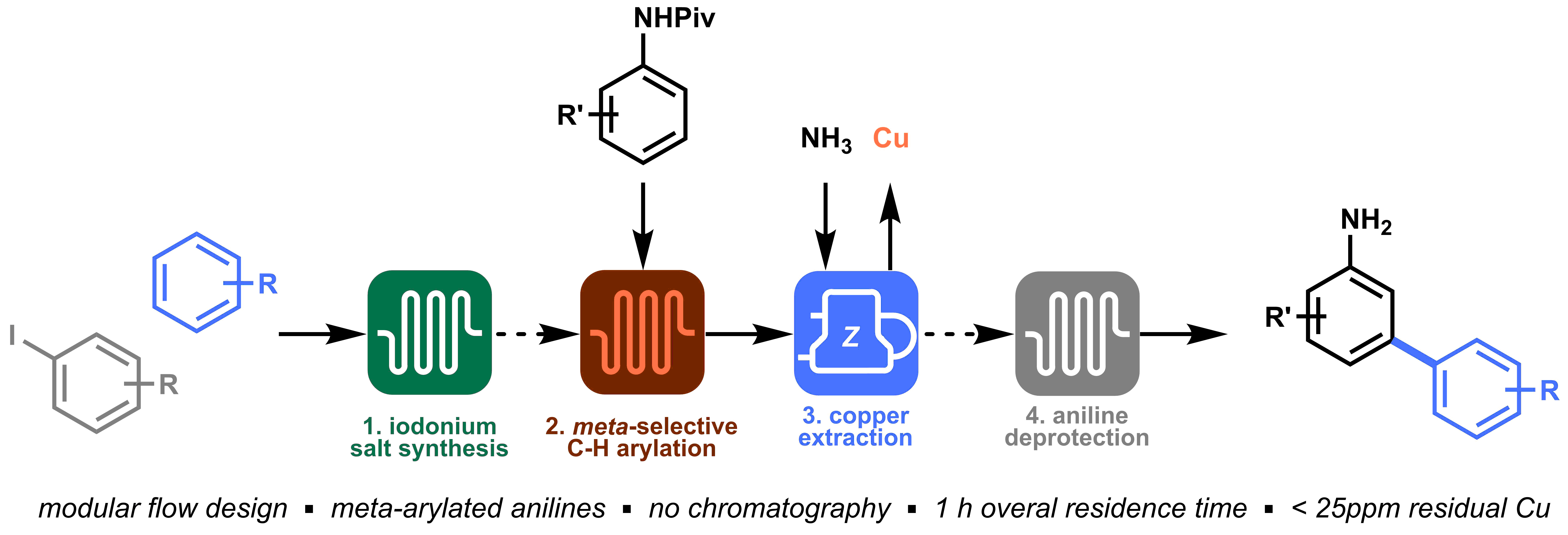
Last but not least, orchestrating all modules in an integrated process allowed to prepare meta-arylated anilines within a total time frame of 1 hour with excellent yield and purity, and without the need of chromatography! (Figure 5).
A final word.
Looking back at this project, I think our winning strategy was the ability to identify the four key elements in the synthesis of meta-arylated anilines, reevaluate and improve them in order to obtain a final orchestrated process. The four separate modules that we developed (i.e. diaryliodonium synthesis, meta-selective C−H arylation, inline copper extraction and aniline deprotection) allowed us to directly access meta-arylated anilines in a time-efficient manner. The success of this project is, of course, the results of a great team effort, and it was my pleasure to share the struggles and satisfactions of this work with Gabriele, Kirsten, and Timothy.
We hope that each of the developed modules will be of high value and find widespread use in the synthesis of drugs, natural products and other molecules of relevance for the chemical industry.
As a group, and thanks to incentive funding from the Netherlands Organization for Scientific Research (NWO), we believe in the importance of open access scientific publications, which is why this manuscript is available free of charge for everybody who is interested!
Enjoy the reading!
Ciao,
The paper discussed in this blog was published as: A Modular Flow Design for the Meta-selective Arylation of Anilines. H. P. L. Gemoets, G. Laudadio, K. Verstraete, V. Hessel, T. Noël, Angew. Chem. Int. Ed. DOI: 10.1002/anie.201703369.
References :
[1] R. J. Phipps, M. J. Gaunt, Science 2009, 323, 1593-1597.
[2] B. Chen, X. L. Hou, Y. X. Li, Y. D. Wu, J. Am. Chem. Soc. 2011, 133, 7668-7671.
[3] Y. Zhang, T. F. Jamison, S. Patel, N. Mainolfi, Org. Lett. 2011, 13, 280-283.
[4] European Medicines Agency (EMEA), “Guidelines on the specification limits for residues of metal catalysts or metal reagents”, can be found under http://www.ema.europa.eu/ema/, 2008.