Frustration as a source of inspiration
I am pretty sure any PI can name a few experiments which their students detest. Not because they are useless (despite the often-used bluff feedback that it is “not absolutely necessary” for the paper), but because these experiments are tedious, time-consuming and frustrating. Amongst the most hated experiments, one could easily place measuring IR spectra (Such an old technique, who needs that anyway?), distilling reagents and solvents (Can’t we just buy a new bottle?), titrations (Do you not believe the concentration written on the bottle?), … In our lab, and probably in many other ones, a special place on that ranking is occupied by Stern-Volmer analysis.
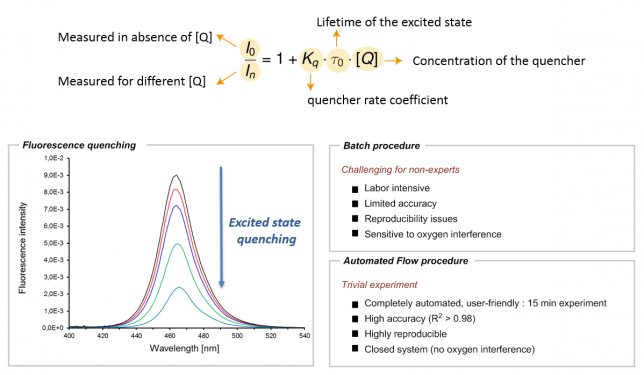
Those of you working on photoredox catalysis probably know too well what I am talking about. For those who don’t, here’s a brief explanation: once in their excited state, photocatalysts tend to return to their ground state by dissipating the energy in the form of emitted light (also called fluorescence). However, in the presence of appropriate organic molecules (often referred to as quenchers), the excited photocatalyst can transfer that excited state energy to a suitable quencher via a single electron or energy transfer. As a result of this interaction, a reduction in the fluorescence of the photocatalyst can be observed (Figure 1). The kinetics of this phenomenon can be explained by the Stern-Volmer relationship, thus making this experiment crucial to elucidate which molecule(s) can interact with the photocatalyst and at which rate they can do so. In other words, the Stern-Volmer relationship provides valuable information in the elucidation of any photocatalytic reaction mechanism.
However, as for all kinetic studies, Stern-Volmer kinetics is not that easy (Figure 1). The process is labor-intensive and sensitive to oxygen, thus often making it difficult to obtain quantitative data. In our own experience, we noticed that, depending on the students’ experience (BSc, MSc, Ph.D. or postdoc level), we usually obtained good qualitative results but the reproducibility and accuracy of the data were often problematic. Since we strive to publish the best achievable data, this eventually resulted in the necessity to repeat the experiment several times, sometimes even by several different students. Talking with other colleagues in the field, we realized this was somewhat a general issue in many groups. In some labs, they even attempted to fix this problem by appointing one student as the Stern-Volmer expert, in order to avoid wasting too much time.
As frustration is typically a good reason to initiate change, we felt that an automated tool to conduct Stern-Vomer analysis would be extremely useful (Figure 1). It would not only greatly reduce valuable experimental time but it would also make sure that the process can be carried out with high precision and reproducibility by basically any chemist, regardless of their experience. Automated platforms have become increasingly important and have often been combined with flow chemistry to assist chemists in their synthetic efforts. Seminal works by the groups of Jensen (MIT), Bourne (Leeds), Ley (Cambridge), Rueping (RWTH) and many others have paved the path for a future in which a machine-assisted approach towards organic synthesis allows chemists to disregard the routine aspects of their laboratory work.[1] Indeed, we felt that Stern-Volmer and other quenching studies were requiring too much of our precious research time.
The kick-off of our automation activities
At that point, we had little to no experience with the automation of chemical processes. However, at TU Eindhoven, we are lucky to have every year bright Chemical Engineering MSc students, who are motivated to take on important challenges within their graduation thesis. Under Koen Kuijpers and Dario Cambie’s supervision, Koen Drummen (having too many Koens at the same time in the lab was also confusing for us, so we started calling them Koen2 for brevity J) started his thesis in September 2016 working on the project. He took our UV-VIS spectrophotometer (Avaspec-2048) and equipped it with a 100 μl quartz flow cuvette which allows measuring the fluorescence of the photocatalyst (Figure 2). Inspired by the Glorius mechanism-based luminescence screening method,[2] we also envisioned to carry out automated screening experiments of various quenchers. For this reason, Koen Drummen also installed an autosampler which allowed us to inject different quenchers sequentially into the continuous-flow stream.
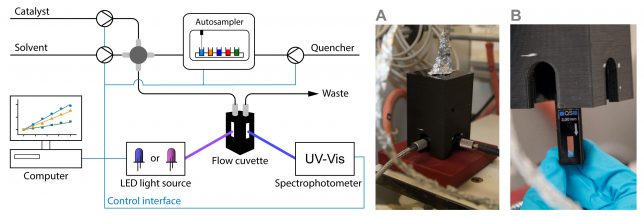
Installing and connecting all the different units together was relatively straightforward. The real confrontation, in this case, was to write a suitable code to make sure all the different units can talk to each other. Hereto, we had to learn Python. I still remember entering the office and seeing Koen2 watching YouTube videos to solve all their issues with the code. Who doesn’t love a nice YouTube tutorial? We partly have to thank some YouTubers: in the end, Koen2 fixed the code and started to run the first automated fluorescence quenching screenings.
Next, we also developed a graphical user interface which allows you to rapidly fill in the compounds to be screened (Figure 3). This makes the entire automated setup really intuitive, basically making it possible for anyone to perform Stern-Volmer quenching experiments, regardless of their ability to code. A cool feature of our software is that, once the analysis is finished, the results are automatically sent by email as a pdf file. Easy enough, right?
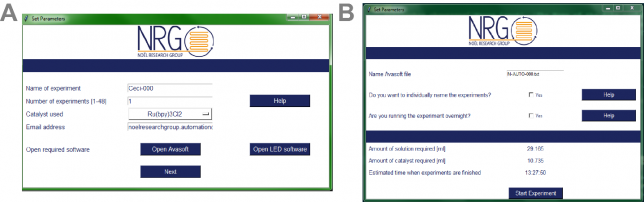
At that point (we are now early 2017), another MSc student, Niels Koenig, joined our group eager to further improve the software. He added a new feature to the software, namely the possibility to automatically color-code the results obtained in the quenching experiments. If a compound was found to be a strong quencher (> 50% quenching efficiency), a green color was assigned to its fluorescence peak while a weak quencher (< 25% efficiency) was assigned a red color. This feature gives in one blink of an eye the confirmation on which quenchers are interesting to test in further reactions and which ones can be discarded.
Time to test the automated platform.
Once our engineers had a working platform ready, it was time to put it to the test. At this point, I asked our photocatalysis expert Cecilia Bottecchia to collaborate with Koen Kuijpers to see how we could take advantage of the technology. The first thing we did was a Stern-Volmer analysis to determine the rate at which TMEDA can quench the excited state of 9-mesityl-10-methyl acridinium perchlorate (Mes-Acr+) (Figure 4). By adding in flow increasing concentrations of quencher, a progressive decrease of the fluorescence emission intensity can be observed. Next, the software converts the obtained data into the classical linear Stern-Volmer profile. From the slope of the line, one can now easily obtain the quenching kinetic constant for a certain quencher. Notably, ten consecutive experiments demonstrated that the data produced by our automated platform was consistent and reproducible, with exactly the same slope (and thus quenching kinetic constant) in each experiment! What a difference compared to our laborious manual procedure!
With the optimized system, we could now run a single Stern-Volmer experiment within only 15 minutes. The only thing you need to do is to prepare the photocatalyst and the quencher solutions. After that, it’s all about hitting the start button, sit back, relax (or work on something else :)) and enjoy your results.
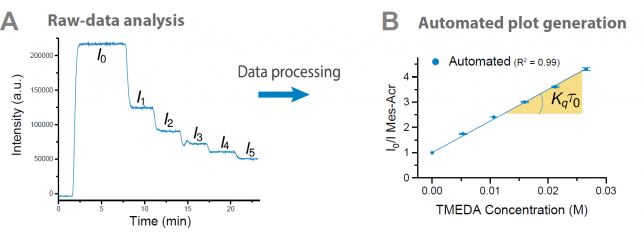
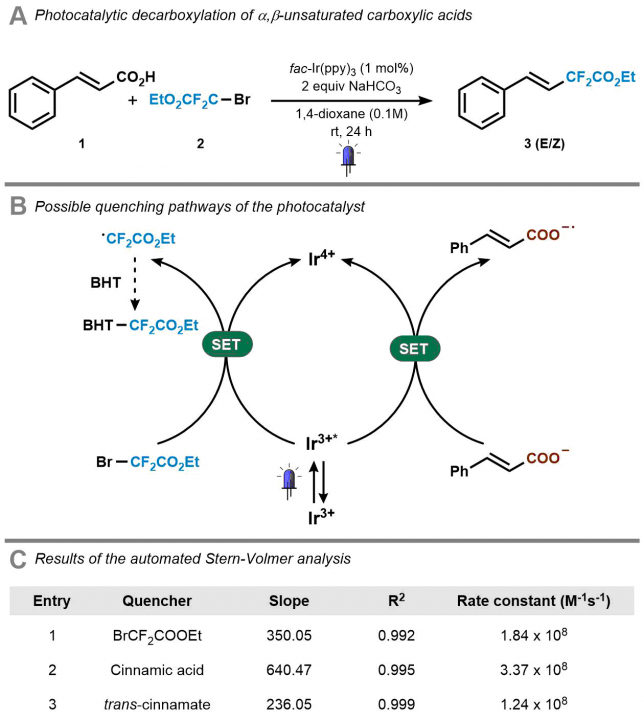
Next, we investigated several case studies to validate the screening option of our platform. One specific example that we carried out was the mechanistic investigation of the Ir(ppy)3-catalyzed photocatalytic decarboxylation of cinnamic acids developed by Xiao-Jing Wei in our group.[3] Cecilia prepared the stock solutions and found rapidly that the software and hardware could rapidly generate the data which was required to elucidate the mechanism (Figure 5). It should be noted that in all cases, we obtained high-quality data with R2 >0.99. Now, in one single hour, we obtained both qualitative and quantitative data, while previously we had to struggle sometimes days to get the quantitative data. It might still be true that, as one referee pointed out, “a good photochemist can run a Stern-Volmer analysis in 30 minutes” but we still believe that you will have to work really hard to get everything done in this timeframe. We did some other screening procedures with our system as well but we invite you to have a look at our paper for all the experiments we did. The bottom-line is that such experiments have become a piece of cake making it actually fun to do.
Outlook
Automating luminescence quenching studies has been one of those projects that really changed the daily life in our group. Before we had access to this platform, students hated me when I asked them to do this experiment. One even said (not a joke): “I’d rather add ten more substrates to the scope instead of doing the Stern-Volmer studies!”. While I could not be bribed (I asked for both the substrates and the SV analysis :)), I still felt that it was crucial to at least alleviate the unnecessary issues that these experiments bring along. We also like the story behind this project because it clearly shows how certain ideas take shape within our group: we wanted to develop some novel synthetic methods via visible light photoredox catalysis, but we ended up with a creative solution to overcome technical hurdles along the way. Simply put, we found a mean to our end. One anonymous referee during the peer-review process really made us proud: “This article presents a valuable and practical advancement for the field of photoredox catalysis. […] Overall, the development of an automated flow platform that significantly accelerates the collection and processing of fluorescence quenching data in a time and labor-efficient manner will be immediately impactful to the organic community.” Our system was and still is impactful in our group and we all hope it will be for your work as well! Don’t hesitate to contact us if you want to give it a shot!

See you,
Tim, Cecilia and Koen
The paper discussed in this blog was published as: A fully automated continuous-flow platform for fluorescence quenching studies and Stern-Volmer analysis. K. P. L. Kuijpers, C. Bottecchia, D. Cambie, K. Drummen, N. Koenig, T. Noël, Angew. Chem. Int. Ed. DOI: 10.1002/anie.201805632.
References:
[1] (a) D. E. Fitzpatrick, C. Battilocchio, S. V. Ley, Enabling Technologies for the Future of Chemical Synthesis. ACS Cent. Sci. 2016, 2, 131-138. (b) B. J. Reizman, K. F. Jensen, Feedback in Flow for Accelerated Reaction Development. Acc. Chem. Res. 2016, 49, 1786-1796.
[2] M. N. Hopkinson, A. Gomez-Suarez, M. Tederes B. Sahoo, F. Glorius, Accelerated Discovery in Photocatalysis using a Mechanism‐Based Screening Method. Angew. Chem. Int. Ed. 2016, 55, 4361-4366.
[3] X.-J. Wei, W. Boon, V. Hessel, T. Noel, Photocatalytic Decarboxylation of α,β-Unsaturated Carboxylic Acids: Facile access to Stereoselective Difluoromethylated Styrenes in Batch and Flow. ACS Catalysis 2017, 7, 7136–7140.







 .
.







 A new book on “Organometallic Flow Chemistry” with Tim as editor has been published in the series “Topics in Organometallic Chemistry” and is available, in hardcover and eBook, on
A new book on “Organometallic Flow Chemistry” with Tim as editor has been published in the series “Topics in Organometallic Chemistry” and is available, in hardcover and eBook, on