Our most recent paper dealing with the electrochemical synthesis of sulfonamides was just published in Journal of American Chemical Society (DOI: 10.1021/jacs.9b02266). For our group, it was the first paper in this prestigious journal and we are particularly proud of the work itself. It has been an interesting project which required a lot of hard work by many co-authors and we are happy to take you today behind the scenes and providing you with some insights in the discovery process.
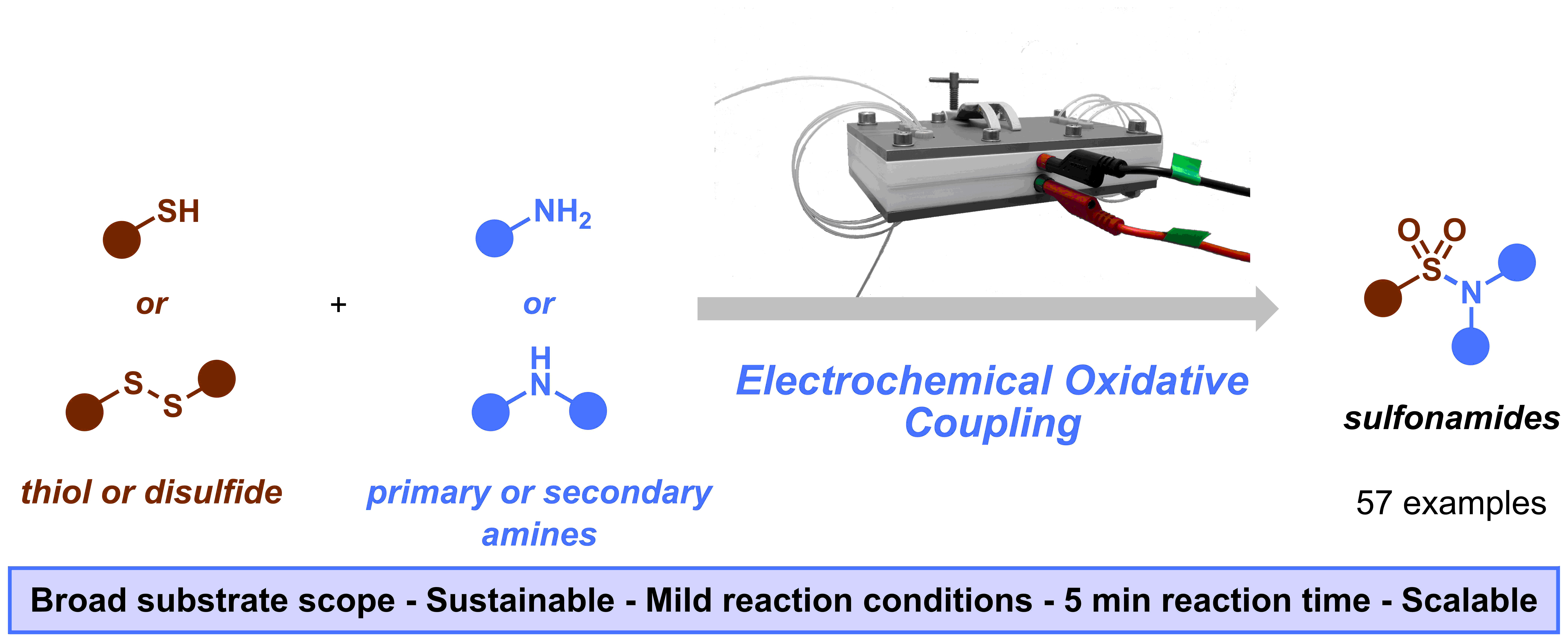 Figure 1. Graphical abstract of our “Sulfonamide synthesis through electrochemical oxidative coupling of amines and thiols”.
Figure 1. Graphical abstract of our “Sulfonamide synthesis through electrochemical oxidative coupling of amines and thiols”.
In search of inspiration
Our group is mostly known for its work on flow photochemistry but about three years ago, we started getting interested in electrochemistry. For us, electrochemistry seemed like a complementary activation mode compared to photoredox catalysis, both allowing to generate synthetically useful radicals. In addition, electrochemical setups are often affected by process-related problems, like mass- and heat-transfer limitations, which was for us the proof that a technological solution would of great added value.
After our first paper on the electrochemical oxidation of thioethers and thiols,1 we decided to design a new electrochemical flow microreactor to carry out our chemistry in a more reliable and scalable way.2 In the meantime, we started thinking about new electrochemical transformations that we could develop. The group’s aim is to develop new, useful synthetic methodology and utilize cutting-edge technology to give a further boost to the chemistry. At that time, our experience on electrochemistry was still very rudimentary and we were reading a lot of different publications to get a bit an image of the field. Ultimately, we found an amazing review entitled “Nonaromatic Aminium Radicals”.3 After reading this review carefully, we had an idea: what if we try to make sulfenamides electrochemically and then further oxidize these species to make sulfonamides? We had already some expertise with the oxidation of thioethers and if we could combine both the S–N coupling with the subsequent oxidation of the sulfur, we had a great method that allowed us to activate commodity chemicals, such as thiols and amines. Moreover, sulfonamides are interesting moieties which are quite common in pharmaceuticals and agrochemicals. Recently, a couple of interesting methods to prepare or functionalize sulfonamides appeared in the literature, incl. the work of Willis4 and MacMillan.5 Taken all of this information together, we had the feeling that this project would be both challenging and of high interest to the synthetic community.
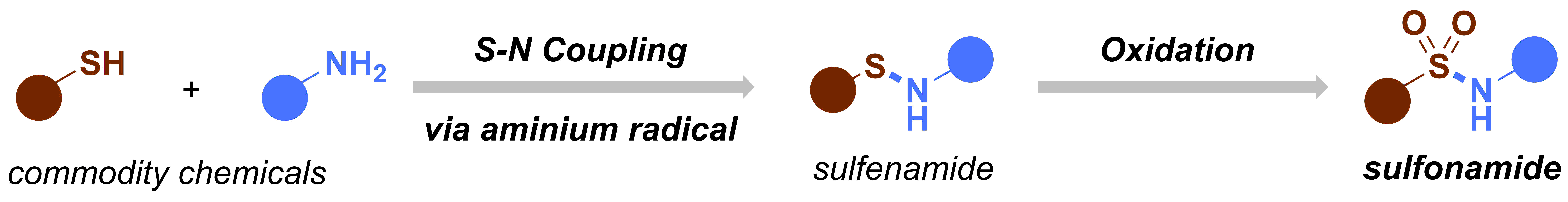
Figure 2. Original idea for the synthesis of sulfonamides.
A working reaction = A wedding present for Gabriele
At that time, Lisa Struik joined our group for her master thesis. Initially, she worked on the experimental validation of our electrochemical flow reactor.2 When that project was finalized, we suggested her to try this idea we had on the synthesis of sulfonamides. Eager to work on this new project, she started with the experiments. One of the first things she said was the following: “We have a huge problem: the reaction is forming a solid and crashes out in pure acetonitrile. I can NOT carry this out in flow!” Our answer to this was “It is probably just the acid-base adduct, just add a bit of water and it will be fine”. Indeed that solved the issue pretty well and not only that, it really boosted the reactivity significantly. To our delight, the results were almost immediately perfect. You cannot imagine how excited we were when we saw that the major product was not the sulfenamide, but the sulfonamide directly!!! This gave us the confidence to increase our research efforts: We started right away with the further optimization and got some help from Christiane Schotten. Christiane was a visiting PhD student coming from the Browne group at Cardiff University and she helped us to test different reaction conditions, residence times, mediators and acids.
Once we had the optimal reaction conditions in hand, Christiane and Lisa started working on the substrate scope. Gabriele had to take a short break at that point as he got married and went on honeymoon on a cruise. Gabriele still remembers that every time they went on land he was anxious to check his emails to know if there was any progress. Upon his return, we had to say goodbye to Christiane and Lisa and for a while, Gabriele had to work alone on the scope. Luckily, Efstathios Barmpoutsis (AKA Stathis), another master student, joined the project, rolled up his sleeves and got immediately to work. It was at that point that we realized how crucial it was to clean the electrodes thoroughly in between reactions. If you don’t do that, yields drop gradually and the results are difficult to reproduce. However, with the proper cleaning, as we described in detail in the Supporting Information of our manuscript, this issue is completely overcome and highly reproducible results are obtained. A little bit later another issue arose: heterocyclic arenes were completely unreactive under our reaction conditions (Figure 3). After some optimization of the reaction conditions, we found that the reaction could be carried out when 1 equiv. of pyridine was added to the reaction mixture. We surmise that pyridine functions mainly as an electron mediator. To be sure, we also evaluated whether its presence was required in the other reactions we already screened but there it did not prove to be essential.
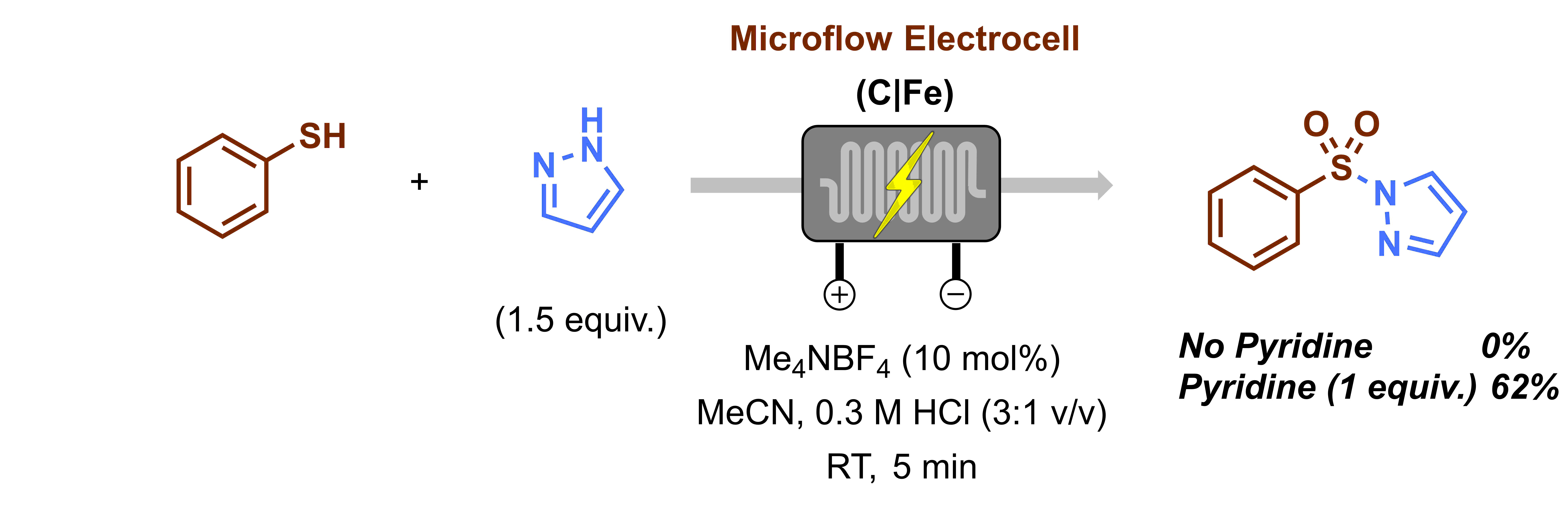
Figure 3. Optimization of heteroaromatic compounds
While Stathis and Gabriele were working on the reaction scope, an Erasmus student Sebastian Govaerts joined the team. (Basically, he returned to our group as he previously worked with Gabriele on the sp3 C–H oxidation project.6) He focused mainly on the batch scope as we wanted a thorough comparison with the results obtained in flow. In most cases, the yields were lower in batch and the reaction required 24 h and 100 mol% of supporting electrolyte to reach full conversion. In flow, the reaction was done within 5 min and needed only 10 mol% of supporting electrolyte. However, batch does have its advantages. For those reactions that were very slow or resulted in precipitate formation, we preferred the batch procedure.


Figure 4. (Left) Electrochemical batch reactor; (Right) Electrochemical flow reactor
With the finish line in sight – What is the mechanism?
Within sight of the finish line, it was time to gain some mechanistic understanding. Kinetic experiments revealed that the thiol dimerizes within 20 seconds to the corresponding disulfide. This observation indicates that some of the most odorous thiols can be replaced by their corresponding disulfide, a feature most chemists will be happy about. Next, Gabriele started with some quenching experiments and he was able to rapidly trap the aminium radical. With that preliminary result in hand, we were almost sure that the sulfenamide was one of the intermediates in our transformation. Indeed, we were able to isolate some sulfenamide and when subjected to our reaction conditions, the targeted sulfonamide was obtained in quantitative yield.
Finally, we had to do some cyclic voltammetry. Using a potentiostat, Gabriele recorded spectra of all relevant starting materials, including amines, thiols and disulfides. Despite some hints, it proved to be not enough evidence to pinpoint the entire mechanism. Then he decided to “titrate” thiophenol with cyclohexylamine and recorded different spectra. We firmly believe in the famous Latin maxim “Fortuna audaces iuvat”, which means literally “fortune favours the bold”. Indeed, at a certain moment, we found some white precipitate in the cell. It was a Friday evening, around 8 pm. Gabriele filtered the precipitate and ran to the NMR: indeed, it was the cyclohexylammonium thiophenolate (Figure 5), as was also observed by Lisa. And, when we tried to record the cyclic voltammogram of that compound, we knew that we had finally found the missing piece (Figure 6). We still carried out some additional control experiments to be 100% sure (you can find them in the supporting information of the paper) but from that moment on we knew the work in the lab was basically done.
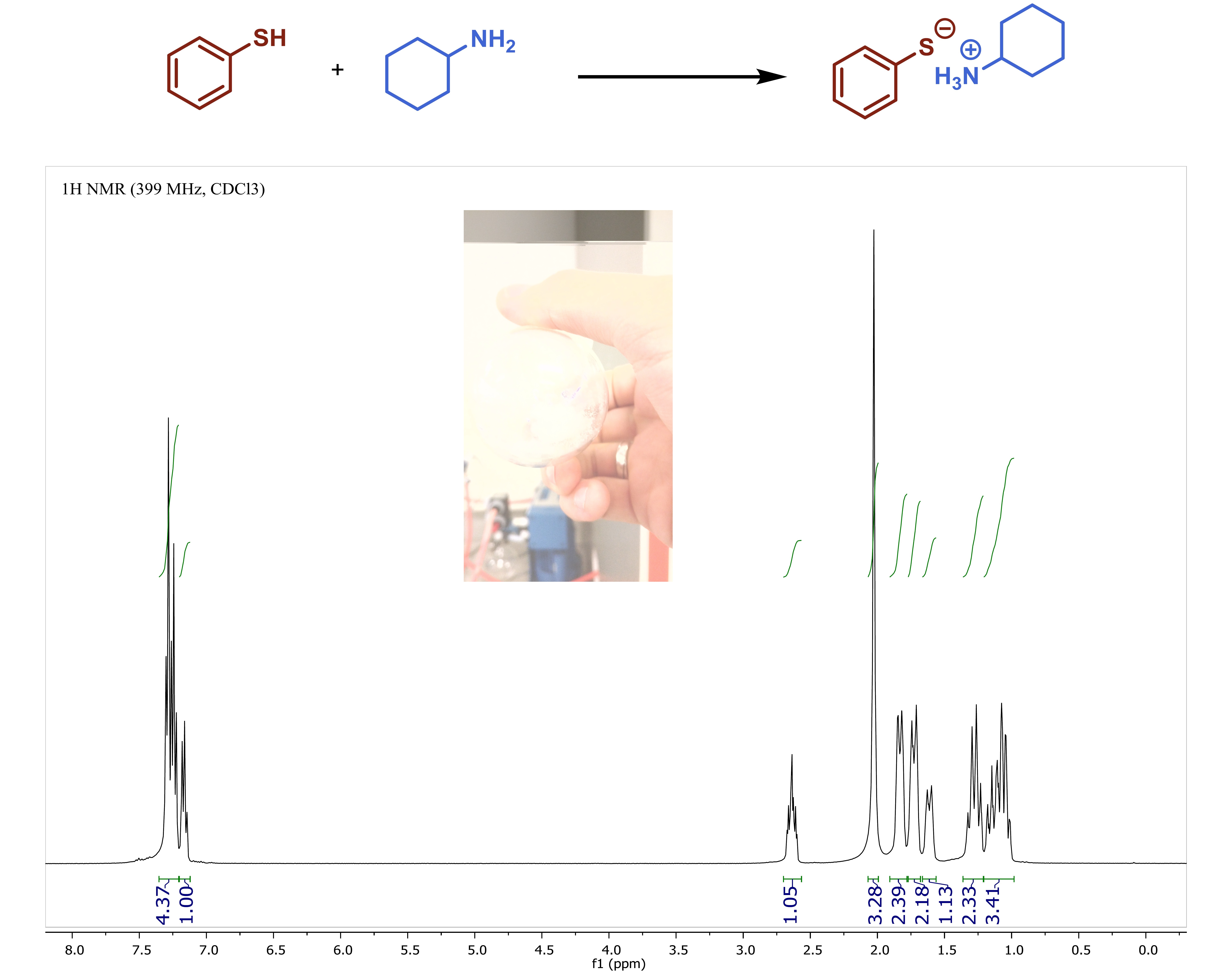
Figure 5. Picture of the white solid and its 1H NMR spectrum
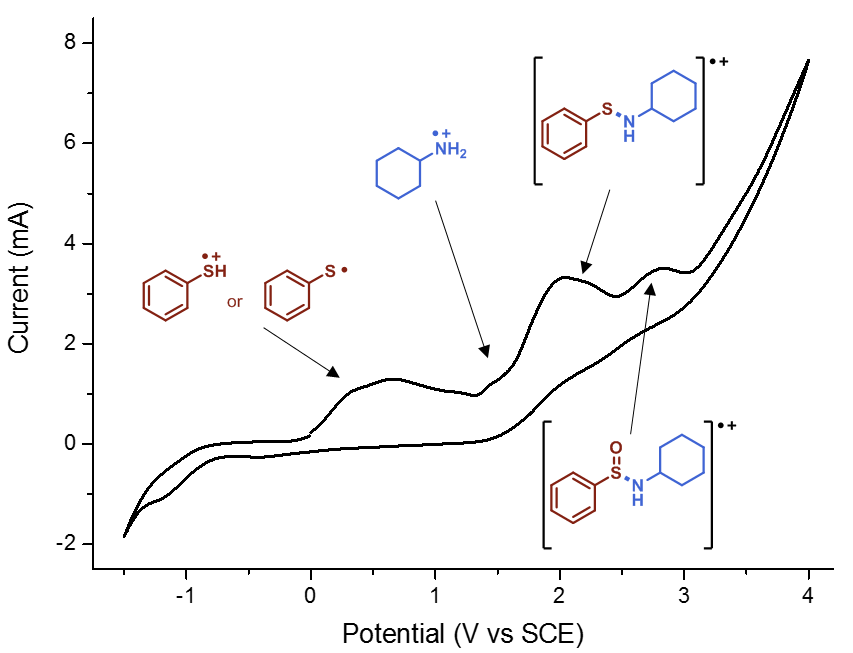
Figure 6. CV of cyclohexylammonium thiophenolate
A final word
The possibility to obtain complex moieties, such as sulfonamides, simply by stitching two commodity chemicals together using electrons is, in our opinion, a key feature of our electrochemical methodology. Moreover, we saw that microflow technology really made a difference in this transformation providing the experimentalist with practical reaction conditions and operational flexibility to rapidly investigate the reaction scope. This can be attributed to the small interelectrode gap (250 μm), the high mass transfer to the electrodes and the large electrode surface to volume ratio.
Currently, we are working hard to finish some more electrochemical methods of which we are really excited. So stay tuned and keep an eye on our group website and twitter accounts.
Ciao,
Gabriele and Tim
The paper discussed in this blog was published as “Sulfonamide synthesis through electrochemical oxidative coupling of amines and thiols”. by Gabriele Laudadio, Efstathios Barmpoutsis, Christiane Schotten, Lisa Struik, Sebastian Govaerts, Duncan L. Browne, and Timothy Noël, Journal of American Chemical Society 2019, DOI: 10.1021/jacs.9b02266
References:
[1] Laudadio, G.; Straathof, N. J. W.; Lanting, M. D.; Knoops, B.; Hessel, V.; Noël, T. Green Chem. 2017, 19 (17), 4061–4066.
[2] Laudadio, G.; de Smet, W.; Struik, L.; Cao, Y.; Noël, T. J. Flow Chem. 2018, 8 (3–4), 157–165.
[3] Chow, Y. L.; Danen, W. C.; Nelsen, S. F.; Rosenblatt, D. H. Chem. Rev. 1978, 78 (3), 243–274.
[4] Chen, Y.; Murray, P. R. D.; Davies, A. T.; Willis, M. C. J. Am. Chem. Soc. 2018, 140, 8781-8787.
[5] Kim, T.; McCarver, S. J.; Lee, C.; MacMillan, D. W. C. Angew. Chem. Int. Ed. 2018, 57, 3488-3492.
[6] Laudadio, G.; Govaerts, S.; Wang, Y.; Ravelli, D.; Koolman, H. F.; Fagnoni, M.; Djuric, S. W.; Noël, T. Angew. Chem. Int. Ed. 2018, 57, 4078-4082.